袁豆豆,周秀琪,庞雪晴,杜家艳,周萍萍
*(扬州大学 生物科学与技术学院,江苏 扬州,225009)
*通信作者 周萍萍,副教授,E-mail:ppzhou@yzu.edu.cn
摘要 咖啡酸是一种天然酚酸类化合物,具有抗氧化、抗肿瘤、抗炎等多种生物学活性,咖啡酸也是迷迭香酸、绿原酸和咖啡酸苯乙酯等高附加值化合物合成的重要前体,在医药、食品和化妆品等行业具有较大的应用价值。为实现咖啡 酸 绿 色 可 持 续 生 产 , 在 前 期 构 建 的 产 咖 啡 酸 酿 酒 酵 母 菌 株 中 , 敲 除 苯 丙 氨 酸 途 径 中 的 预 苯 酸 脱 水 酶 基 因(PHA2),将咖啡酸产量提高 42%。而敲除色氨酸途径中邻氨基苯甲酸合成酶基因(TRP2),对咖啡酸积累影响不大。之后,回补了 URA3、HIS3、MET15 营养标记基因,然后将工程菌株在 5 L 的发酵罐中进行了补料分批发酵,使得咖啡酸产量达到了 9.3 g/L,是目前文献中报道微生物发酵生产咖啡酸的最高水平,该研究为咖啡酸的绿色生产以及其衍生物的高效微生物合成奠定了基础。
关键词 酿酒酵母;咖啡酸;预苯酸脱水酶;邻氨基苯甲酸合成酶;生物合成
Abstract Caffeic acid is a natural phenolic acid with various pharmaceutical properties, including antioxidant, anti -tumor and anti-inflammatory effects. It is also an important precursor for biosynthesis of other valuable compounds such as rosmarinic acid, chlorogenic acid and phenethyl caffeate. Therefore, caffeic acid has significant value in pharmaceutical, fo od, cosmetic and other industries. To achieve green and sustainable production of caffeic acid, the prephenate dehydratase gene (PHA2), which is involved in the biosynthesis of phenylalanine, was knocked out in a previously constructed caffeic acid produced Saccharomyces cerevisiae, resulting in a 42% improvement in caffeic acid production. Additionally, the anthranilate synthase gene (TRP2), which is responsible for tryptophan biosynthesis, was deleted. However, this had little effect on coffee acid production. The engineered strain was further complemented with auxotrophic marker URA3, HIS3, MET15. In a subsequent fed-batch fermentation process conducted in a 5 L bioreactor, the engineered strain achieved a caffeic acid production of 9.3 g/L, this is currently the highest reported titer of caffeic acid produced by engineered microbial cells. T his study provides a foundation for the green production of caffeic acid and its derivatives.
Key words Saccharomyces cerevisiae; caffeic acid; prephenate dehydratase; anthranilate synthase; biosynthesis
咖啡酸(caffeic acid),又称 3, 4-二羟基肉桂酸,是存在于多种植物中的一种天然酚酸类化合物,具有抗氧化、抗炎、抗肿瘤等多种生物学活性[1-3]。此外,咖啡酸是绿原酸[4]、迷迭香酸[5]、丹酚酸 B[6]、咖啡酸苯乙酯[7]等高附加值化合物的前体,在食品、药品和化妆品等领域具有广泛的应用[8]。由于咖啡酸存在于咖啡以及茵陈、菜蓟、金银花等多种植物中,因此,可以从这些植物中直接提取来制备。但是植物生长周期长、产物积累量低,且在植物提取过程中需要用到多种溶媒,限制了其大规模的生产。咖啡酸也可以由苯酚或取代苯酚经过化学催化剂反应生成,但化学合成过程中反应条件要求高、制备工艺复杂、成本高,并不是一条绿色经济的生产路线。随着合成生物技术的发展,代谢改造微生物合成咖啡酸成为一种有效的替代方法。
在植物中,苯丙氨酸解氨酶(phenylalamine ammonia lyase,PAL)催化 L-苯丙氨酸脱氨生成反式肉桂酸,反式肉桂酸在肉桂酸-4-羟化酶(4H)催化下生成对香豆酸,最后对香豆酸在对香豆酸-3-羟化酶(C3H)催化下生成咖啡酸。植物来源的 C3H 和 C4H 属于 P450 酶,在微生物中难以高效表达[9]。BERNER 等首次报告了西班牙糖丝菌(Saccharothrix espanaensis)来源的 4-香豆酸-3-羟基化酶(由 Sam5 编码)和酪氨酸解氨酶(由Sam8 编码),在弗氏链霉菌中表达可以将 L-酪氨酸转化为咖啡酸[10]。
CHOI 等将这两个来源于西班牙糖丝菌的 Sam8 和 Sam5 基因在大肠杆菌表达,成功将对香豆酸转化为咖啡酸[11]。LIN 等发现来自大肠杆菌自身的 4-羟基苯乙酸 3-加氧酶(Hpa B)和 4-羟基苯乙酸 3-还原酶(Hpa C)可以替换微生物中难表达的 C3H。通过将大肠杆菌来源的 Hpa BC 与荚膜红细菌(Rhodobacter capsulatus)来源的酪氨酸解氨酶(tyrosine ammonia-lyase,TAL)在大肠杆菌中共表达,成功构建出了一条咖啡酸异源合成途径,并通过解除酪氨酸的反馈抑制步骤,获得了 50.2 mg/L的咖啡酸[12]。进一步敲除大肠杆菌中苯丙氨酸合成途径以及系统优化酪氨酸合成途径等,使得咖啡酸的产量达到了 766.7 mg/L[13]。2022 年,研究人员利用大肠杆菌为出发菌株,表达了约氏黄杆菌(Flavobacterium johnsoniaeu)来源的 Fj TAL 和大肠杆菌内源的 Hpa BC,并通过优化基因拷贝数、敲除酪氨酸合成的竞争途径、强化辅因子 FAD 的合成以及过表达转运蛋白 ycj P,使得咖啡酸摇瓶产量达到了 775.7 mg/L,通过在 5 L 发酵罐进行发酵条件优化后,发酵 66 h 咖啡酸产量达到了 7.9 g/L[14],这是目前报道的微生物合成咖啡酸的最高产量。在上述构建的大肠杆菌工程菌中,代谢途径中的基因常常通过多个游离质粒进行表达,菌株稳定性差,在培养过程中需要添加多种抗生素,且培养过程中需要添加异丙基-β-D-硫代半乳糖苷(isopropyl-beta-D-thiogalactopyranoside,IPTG)进行诱导,影响了后期的提取工艺,不具备食品安全性和生产经济性。
酿酒酵母作为基因操作简单的真核微生物,与大肠杆菌相比,不易染噬菌体,且在生物安全性方面和植物基因的功能表达方面有其独特的优势,是植物天然产物异源合成的极佳宿主。在酿酒酵母中,LIU 等筛选了不同物种来源的 Hpa B 和 Hpa C,并通过组合表达发现铜绿假单胞菌的 Hpa B 和肠沙门氏菌的 Hpa C 的组合效果最佳,可以有效地将对香豆酸转化到咖啡酸。并结合圆红冬孢酵母(Rhodotorula toruloides)来源的 Rt TAL 表达,在酿酒酵母中实现了咖啡酸的从头合成,产量达到289.4 mg/L[15]。2022 年,CHEN 等通过改造中心代谢提高 NADPH 供应,通过构建胞浆 FAD(H2)合成途径以及将线粒体 FAD(H2)导到胞浆以提高 FAD(H2)供应,显著提高了咖啡酸生物合成效率,摇瓶产量达到了 1.6 g/L,在 5 L发酵罐中补料分批发酵产量达到了 5.5 g/L[16]。
2021 年,本课题组以酿酒酵母内源 L-酪氨酸为前体,将粘红酵母(Rhodotorula glutinis)来源的Rg TAL 基因、铜绿假单胞菌来源的 Hpa B 基因和沙门氏菌来源的 Hpa C 基因组合,在酿酒酵母中构建了一条咖啡酸生物合成途径[17],并通过敲除苯丙氨酸反馈抑制的 Aro3、过表达酪氨酸反馈抑制不敏感突变体 Aro4K229L 和 Aro7G141S、敲除丙酮酸脱羧酶 Aro10和表达运动发酵单胞菌来源的环己二烯脱氢酶 Tyr C(图 1),构建的酿酒酵母菌株 YCA113-8B,其咖啡酸产量提高了约 2.6 倍,摇瓶产量达到了 568.5 mg/L。为了进一步提高咖啡酸合成产量,提高前体供应至关重要。在上述合成过程中,前体分支酸会在邻氨基苯甲酸合酶(TRP2)催化下生成邻氨基苯甲酸,即进入色氨酸支路,而预苯酸则在预苯酸脱水酶(PHA2)的催化下合成苯丙酮酸,而苯丙酮酸会进一步经酶催化转化生成苯丙氨酸,即进入苯丙氨酸途径(图 1)。因此,为使更多的碳代谢流流向咖啡酸合成途径,本研究在前期构建的咖啡酸合成菌株基础上,利用 CRISPR/Cas9 编辑系统敲除 PHA2 和 TRP2 基因,考察了其对酿酒酵母咖啡酸合成的影响。并对工程菌株中导致氨基酸营养缺陷的 URA3、HIS3、MET15 标记基因进行了回补。最后将工程菌株在 5 L 发酵罐中进行了补料分批发酵,以期提高咖啡酸的产量。本研究为发酵生产咖啡酸奠定了基础,也为其他天然酚酸类化合物的生物合成提供了方法参考。
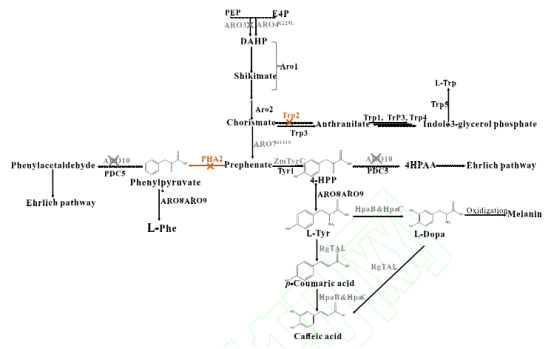
图 1 酿酒酵母中咖啡酸生物合成途径
Fig. 1 The biosynthetic pathway of caffeic acid in Saccharomyces cerevisiae
1 材料与方法
1.1 实验材料
1.1.1 菌株与质粒
研究使用的引物、质粒和菌株分别见表 1~表 3。大肠杆菌(Escherichia coli)DH5α用于重组质粒的克隆和扩增实验。前期构建的酿酒酵母咖啡酸合成菌株 YCA113-8B 为本实验的出发菌株。菌株、质粒全部保藏在本实验室。
表1 本研究所使用的引物
Table 1 Primers used in this study

表2 本研究所用的质粒
Table 2 Plasmids used in this study

1.1.2 母液与培养基
100×氨基酸母液:L-甲硫氨酸、L-组氨酸、L-色氨酸、L-苯丙氨酸、尿嘧啶质量浓度分别按照2L、2、2、5、2 mg/m L 配制,添加于培养基时按照 1%的体积比。
遗传霉素(G418)母液:按照 20 mg/m L 配制,添加于培养基时按照 1%的体积比。
氨苄青霉素母液:按照 100 mg/m L 配制,添加于培养基时按照 0.1%的体积比。
卡那霉素母液:按照 50 mg/m L 配制,添加于培养基时按照 0.1%的体积比。
Single-straided carrier DNA(ss DNA):20 mg 溶于 10 m L TE Buffer 中,0.22 µm 微孔滤膜过滤除菌,分装后-20 ℃保存。
1 mol/L 醋酸锂溶液:10.2 g 乙酸锂二水定容于 100 m L 蒸馏水中,121 ℃湿热灭菌 15 min,冷却后于 4 ℃保存。
PEG MV3 350(50% M/V):50 g PEG MV3 350 定容于 100 m L 蒸馏水中,121 ℃湿热灭菌 15 min,冷却后于 4 ℃保存。
Luria-Bertani(LB)液体培养基(g/L):LB 肉汤培养基 25 g,115 ℃灭菌 21 min。
Luria-Bertani(LB)固体培养基(g/L):LB 肉汤琼脂 40 g,115 ℃灭菌 21 min。
Yeast Extract Peptone Dextrose(YPD)液体培养基(g/L):酵母蛋白胨 20,无水葡萄糖 20,酵母浸粉 10,115 ℃灭菌 21 min,在配制固体培养基时,添加 1.5%~2%的琼脂。
10×Yeast nitrogen base(10×YNB)母液:YNB 17 g, (NH4)2SO4 50 g,溶于 1 L 蒸馏水中,过滤除菌,添加于培养基时按照 10%的体积比。
Synthetic Defined(SD)固体培养基:葡萄糖 2 g,琼脂粉 1.5 g,蒸馏水 90 m L,115 ℃灭菌 21 min, 使用时应加入 1 m L 100×氨基酸母液和 10 m L 10×YNB 母液。
5-氟乳清酸(5-fluoroorotic acid,Fo A)固体培养基:称取 0.1 g FOA 固体溶于 1 m L 二甲基亚砜中,100 m L SD 固体培养基融化降温至 50 ℃左右,加入 1 m L 100×氨基酸母液、0.1 g/m L Fo A 溶液混匀。
维生素溶液(g/L):生物素 0.05,泛酸钙 1,烟酸 1,肌醇 25,盐酸硫胺素 1,吡哆醇 1,对氨基苯甲酸 0.2,0.22 µm 微孔滤膜过滤除菌,分装后 4 ℃保存。
微量元素溶液(g/L):EDTA 15,Zn SO4·7H2O 10.2,Mn Cl2·4H2O 0.5,Cu SO4 0.5,Co Cl2·6H2O 0.86,Na2Mo O4·2H2O 0.56,Ca Cl2·2H2O 3.84,Fe SO4·7H2O 5.12,115 ℃灭菌 21 min。
分 批 发 酵 补 料 浓 缩 液 Ⅰ(g/L) : 葡 萄 糖 500, KH2PO4 9 , Mg SO4 2.5 ,K2SO4 3.5 , Na2SO 0.28,115 ℃灭菌 21 min。
分批发酵补料浓缩液Ⅱ(g/L):酵母浸粉 250, 115 ℃灭菌 21 min。
2.5 mol/L Na OH 溶液:30 g Na OH 溶于 300 m L 的灭菌水中。
1.1.3 酶与试剂盒
本研究使用的工具酶与试剂盒见表 4。
1.2 实验方法
1.2.1 重组质粒的构建
以质粒 M3-T[18]为模板,分别以 g PHA2-F(Esp3 I)和 t RNA-R(Esp3 I)、g TRP2-F(Esp3 I)和t RNA-R(Esp3 I)为引物,分别扩增得到 PHA2 g RNA-scaffold-t RNA 序列 、 TRP2 g RNA-scaffold-t RNA 序列。使用限制性核酸内切酶 Esp3 I 将扩增所得的基因片段与 TYPE3-M 质粒[18]分别进行酶切,使用 T4 DNA 连接酶将酶切后的基因片段与质粒分别进行连接,导入 E.coli DH5α 感受态细胞中,筛选阳性转化子,分别得到 TYPE3-M- g PHA2 和TYPE3-M-g TRP2 载体。
1.2.2 酿酒酵母重组菌株的构建
以酿酒酵母 BY4741 的基因组为模板,以 HAPHA2-F/HAPHA2-R 为引物,用高保真的 Prime STARTM HS DNA 聚合酶进行 PCR 扩增,获得同源臂 HAPHA2;同理,用 HATRP2-F 和 HATRP2-R引物进行 PCR 扩增,获得同源臂 HATRP2。将构建的质粒载体 TYPE3-M- g PHA2 与同源臂 HAPHA2通过醋酸锂转化法导入 YCA113-8B 酵母菌株中,均匀涂布在 SD+ Ura-平板上。培养 3~4 d 后,挑取酵母转化菌株于 10 μL 无菌水中,从中取 2 μL 至另一含有 8 μL 无菌水的 PCR 管中,100 ℃加热 5 min 后直接用作 PCR 模板。以 PHA2-F 和 HAPHA2-R 引物,利用 KOD One TM PCR Master Mix 聚合酶进行 PCR 扩增,将扩增得到的 PCR 产物进行送测序验证。将测序正确的菌株,名为 YCA113-16B 菌株。为了进行下一轮基因编辑,需要将菌株 YCA113-16B 中含 Cas9 蛋白和 g RNA 的质粒去除。利用在没有筛选压力的培养基中,通过传代,质粒可自然丢失这一原理,将菌株 YCA113-16B 接种于 5 m L YPD 试管中,30 ℃下培养过夜后,再转接一次到 YPD 试管培养过夜。取 1 m L 菌液无菌水水洗两次后稀释 10 000 倍,取 100 μL 涂于 FOA 平板上。若 FOA 板上的单菌落在 SD 不加 URA 试管中不能生长,而在加了 URA 的 SD 试管中能生长,则说明 TYPE3-M- g PHA2 质粒已经去除,菌株命名为YCA113-16B (-)。同理,将 TYPE3-M-g TRP2与同源臂 HATRP2,通过醋酸锂转化法导入 YCA113-16B (-)酵母菌株中,获得 YCA113-20B 菌株。
将 p UMRI-ΔPdc5 用 Sfi I 进行酶切线性化后,通过醋酸锂转化法导入 YCA113-16B (-)酵母菌株中,构建得到菌株 YCA113-23B。为了回补 YCA113-23B 中 HIS3 和 MET15 营养缺陷型基因,以酿酒酵母 BY4742-MO4-His[19]基因组为模板,用 His3-F1/ His3-R1 为引物用高保真的 Prime STARTM HS DNA 聚合酶进行 PCR 扩增,获得 HIS3 营养缺陷型基因。通过醋酸锂转化法,将 HIS3 片段导入到YCA113-23B 菌株中,并将转化细胞涂布在不添加组氨酸而添加了甲硫氨酸和苯丙氨酸的 SD 固体平板上,从而构建出 YCA113-23BH 菌株。以酿酒酵母 BY4742 基因组为模板,用 Met15-F1/ Met15-R1为引物,用高保真的 Prime STARTM HS DNA 聚合酶进行 PCR 扩增,获得 MET15 营养缺陷型筛选标记的基因片段,并将片段导入到 YCA113-23BH 酵母菌株中,将转化细胞涂布在不添加甲硫氨酸但添加了苯丙氨酸的 SD 平板上。从而构建出 YCA113-23BHM 菌株。
1.2.3 重组菌株发酵实验
在摇瓶发酵实验中,将菌株在 YPD 固体培养基中划线,于 30 ℃恒温培养箱中培养 72 h。挑取平板上的菌落接种到含 5 m L YPD 培养基的试管中,30 ℃,220 r/min 培养 15 h,取一定的种子液接至含 50 m L YPD 培养基的 250 m L 摇瓶中,使初始 OD600为 0.05,30 ℃,220 r/min 培养 72 h。
在补料分批发酵试验中,将菌株 YCA113-23BHM 在 YPD 固体培养基中划线,于 30 ℃恒温培养箱中培养 72 h。挑取平板上的菌落接种到含 5 m L YPD 培养基的试管中,30 ℃,220 r/min 培养 15 h,转接 1 m L 菌液至含有 125 m L YPD 培养基的 500 m L 摇瓶中,30 ℃,220 r/min 培养 20 h。将两瓶 250 m L 种子液全部接种到含有2.25 L YPD 培养基的 5 L 发酵罐中,发酵过程初始通气量设定为2.5 L/min,搅拌速率设定为 300 r/min,此时相对溶氧值设定为 100。发酵过程中通过调节搅拌转速和空气流量,将溶氧维持在 20 左右。培养过程中通过自动补加 2.5 mol/L 的氢氧化钠溶液,将 PH 值稳定在 5.5 左右,温度恒定在 30 ℃。发酵初期,每隔 12 h,补 250 g/L 酵母粉溶液 50 m L,直到菌株生长进入平稳期。葡萄糖流加速率在 0~20 m L/h 之间,使发酵液中乙醇含量控制在 5 g/L 以下。
1.2.4 发酵样品处理
发酵结束后,取 600 μL 发酵液,与 600 μL 甲醇超声 15 min,充分混匀。然后,12 000 r/min 离心 1 min,取上清液 100 μL,与 900 μL 50%甲醇-水混匀,用 0.22 μm有机系针式过滤器过滤,用于后续的液相分析。
1.2.5 检测方法
实验中使用 Agilent 1200 高效液相色谱仪,Pntulips QS-C18 plus(4.6 mm×250 mm,5 µm,Puningtech)液相色谱柱,所用流动相为 0.1%甲酸(A 泵)和乙腈(B 泵),流速 1 m L/min,梯度洗脱程序设定如下:0~30 min,A 溶剂 90%~50%,B 溶剂 10%~50%。柱温 35 ℃。每次进样 20 μL,每个样品运行 15 min。咖啡酸的检测波长为 320 nm,出峰时间约为 11 min。
2 结果与分析
2.1 敲除 PHA2 对咖啡酸合成的影响
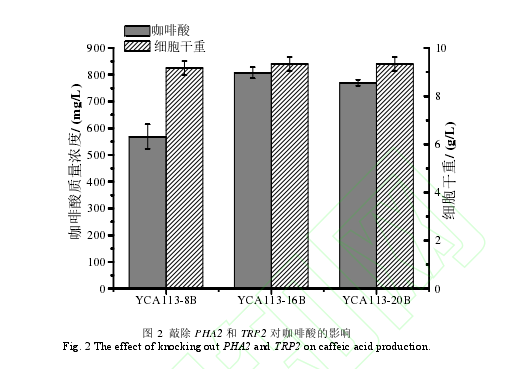
在前期的研究中,以 L-酪氨酸为前体,通过引入咖啡酸合成所需的基因以及敲除苯丙氨酸反馈抑制的 Aro3、过表达酪氨酸反馈抑制不敏感突变体 Aro4K229L 和 Aro7G141S、敲除丙酮酸脱羧酶 Aro10 和表达运动发酵单胞菌来源的环己二烯脱氢酶 Tyr C,构建了一株产咖啡酸的酿酒酵母菌株 YCA113-8B[17]。在该菌株中,L-苯丙氨酸和 L-酪氨酸都是由预苯酸转化形成,L-苯丙氨酸途径是 L-酪氨酸衍生物的主要竞争途径。阻断这条竞争途径可以使得碳代谢流转向 L-酪氨酸衍生物的生物合成。在苯丙氨酸合成途径中,预苯酸脱水酶(PHA2)可以催化预苯酸生成苯丙酮酸,苯丙酮酸在芳香族氨基酸转移酶(Aro8/Aro9)作用下合成苯丙氨酸。为了进一步提高咖啡酸产量,本研究利用 CRISPR/Cas9基因编辑系统敲除了 YCA113-8B 菌株基因组中的 PHA2 基因,构建了 YCA113-16B 菌株。经摇瓶培养 72 h 后,结果如图 2 所示,相比于 YCA113-8B 菌株,咖啡酸的产量提高了 42%,达到了 807.3 mg/L。此外,敲除 PHA2 后,细胞的生物量没有明显变化。结果表明敲除 PHA2 基因能有效地降低前体向苯丙氨酸支路途径的转化,使碳代谢更多地流向咖啡酸合成途径。
2.2 敲除 TRP2 对咖啡酸合成的影响
在酿酒酵母中,由莽草酸途径生成的分支酸,一方面转化生成预苯酸用于 L-苯丙氨酸和 L-酪氨酸的合成,另一方面,会在邻氨基苯甲酸合酶(TRP2)催化下,生成邻氨基苯甲酸,将碳代谢流转向 L-色氨酸合成途径。为了进一步提高咖啡酸的产量,本研究利用 CRISPR/Cas9基因编辑系统进一步将 YCA113-16B 菌株基因组中的 TRP2 基因进行敲除,获得 YCA113-20B 菌株。经摇瓶培养后,敲除 TRP2 基因后,YCA113-20B 菌株咖啡酸产量略微下降(图 2)。之前报道的酵母合成酪醇的研究中,LIU 等发现敲除 TRP2 基因后,酪醇的产量反而下降了 19.7%,其推测是由于敲除 TRP2 基因后,影响了酵母生长所导致的[20]。而在本研究中,敲除 TRP2 后,无论对酵母生物量和咖啡酸产量的影响不是特别明显。推测原因如下:色氨酸分支途径相对较复杂,有 TRP1-TRP5 五个酶通过六步反应将分支酸转化到色氨酸。其中在第一步反应中,除了 TRP2 能催化分支酸转化成邻氨基苯甲酸外,TRP3作为一个双功能的酶,含有谷氨酰胺酰胺转移酶和吲哚 -3-甘油磷酸合酶两个独立的催化结构域,可与 TRP2 协同,催化分支酸向邻氨基苯甲酸的转化[21],因此仅敲除 TRP2 无法完全阻断色氨酸支路。同样地,在 GUO 等的研究中,只敲除 TRP3,酵母菌株生物量和酪醇的产量也无明显变化[22]。因此,在后续的研究中,可以同时敲除 TRP2 和 TRP3,考察其对咖啡酸合成的影响。此外,上述的研究也暗示 TRP2 或者 TRP3 可以作为酵母外源基因整合的位点。
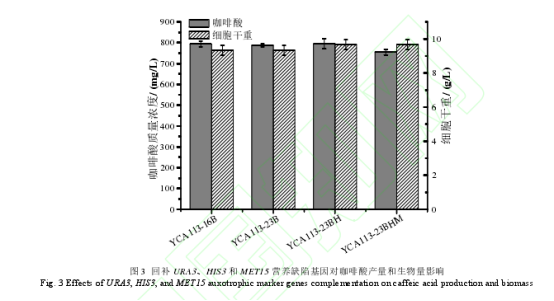
2.3 重组菌株补料分批发酵
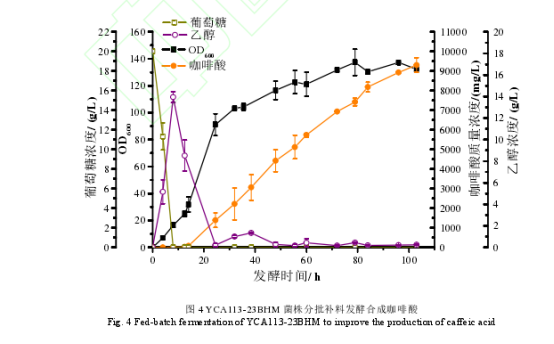
为了进一步提高咖啡酸的产量,计划选用摇瓶中咖啡酸产量相对较高的菌株进行补料分批发酵实验。然而,YCA113-16B 是 HIS3 和 MET15 营养缺陷型菌株,且 YCA113-16B 菌株中,携带有 URA3营养缺陷基因的 TYPE3-M- g PHA2 质粒容易在传代中丢失。为了在补料分批发酵中,避免组氨酸、甲硫氨酸和尿嘧啶缺乏影响细胞生长,首先,在去除了 TYPE3-M-g PHA2质粒的 YCA113-16B(-)菌株中,整合了 URA3 营养标记基因,获得了 URA3 能够稳定遗传的 YCA113-23B 菌株,在此基础上,回补了 HIS3 营养标记基因,获得了 YCA113-23BH 菌株,进一步回补了 MET15 营养标记基因,获得了YCA113-23BHM 菌株。为了考察这些营养缺陷型基因回补后,对菌株咖啡酸和生物量的影响,先进行了摇瓶发酵实验。结果显示,在摇瓶发酵中,进行上述营养缺陷基因回补,对生物量和产量没有造成明显的影响(图 3)。因此,最终选用了 YCA113-23BHM 菌株在 5 L 的发酵罐中进行了补料分批发酵,测试了其生产性能。结果如图 4 所示,在发酵 8 h 左右,葡萄糖耗尽,此时发酵液中乙醇含量达到了13.9 g/L,从 10 h 开始以 1 g/(L·h)的速率补料,12.5 h 时,乙醇浓度下降到了 8.5 g/L,此时,将葡萄糖的补料速度提高到了 2 g/(L·h),菌株进入了对数生长期,发酵 24.5 h后,乙醇的含量只有0.2 g/L。因此将葡萄糖的补料速度提高到了 4 g/(L·h)。但之后,菌株的生长变慢。因此发酵 36 h 之后将补糖速度调为 2 g/(L·h)。发酵 12.5 h 后,咖啡酸快速积累,直到发酵 84 h,咖啡酸的积累速度变缓,葡萄糖补料速度调成了 1 g/(L·h)。经过 103 h 发酵,咖啡酸产量达到了 9.3 g/L,是目前文献中报道的微生物合成咖啡酸的最高产量。但在补料分批发酵中,菌株的生物量较低,OD600最高约为137。因此需进一步优化发酵培养基以及补料工艺,以期提高生物量,从而提高咖啡酸产量。
3 结论与讨论
咖啡酸作为一种重要的活性酚酸类化合物,具有抗氧化、抗癌、抗菌等生物学活性,在食品、医药、化妆品等领域具有广泛的应用。近年来,随着合成生物技术的发展,以大肠杆菌和酿酒酵母为底盘细胞,通过异源代谢途径的表达与优化,已成功实现了咖啡酸的生物合成,但产量仍普遍较低,距工业化应用仍有较大的距离。在前期的研究中,本课题组以酿酒酵母中L-酪氨酸为前体,构建了一株产咖啡酸菌株 YCA113-8B,其摇瓶发酵产量为 568.5 mg/L。为了进一步提高其产量,本研究进一步考察了敲除苯丙氨酸支路途径基因 PHA2、以及色氨酸支路途径基因 TRP2 对咖啡酸合成效率的影响。结果表明敲除 PHA2 基因后咖啡酸的产量提高了 42%。但进一步敲除 TRP2 基因后,咖啡酸产量变化不大。进一步通过回补 URA3、HIS3、MET15 营养标记基因,并在 5 L 生物反应器中进行补料分批发酵,使得酿酒酵母最终积累了 9.3 g/L 咖啡酸,是目前文献中报道的微生物产咖啡酸最高产量。本研究为咖啡酸的高效生物合成提供了方法借鉴,为咖啡酸衍生物的合成提供了优良的底盘细胞。在后续的研究中,为进一步提高咖啡酸产量,可以对莽草酸途径中的两个重要前体磷酸烯醇式丙酮酸(PEP)以及赤藓糖 4-磷酸(E4P)的供应进行改造,以提高咖啡酸前体 L-酪氨酸的代谢流。对香豆酸在 4-羟基苯乙酸-3-单加氧酶(Hpa B)和黄素还原酶(Hpa C)催化下合成咖啡酸。但在表达 Hpa B 和Hpa C 后,发酵液明显呈现棕色。很多研究文献表明 L-酪氨酸也会在 Hpa B 和 Hpa C 的催化下直接生成 L-多巴,L-多巴经氧化生成黑色素[17],从而造成了部分碳代谢流的损失。为此,可以对 Hpa B 和Hpa C 的表达时间进行调控,在对香豆酸积累后,再开启 Hpa B 和 Hpa C 的表达,或者对 Hpa B 和Hpa C 的催化底物专一性进行改造。此外,在补料分批发酵中,需进一步对基础培养基、补料策略进行优化,提高菌株的生物量,从而提高咖啡酸产量。